1. Đặt vấn đề
- Đường kính hẹp của đường thở nhỏ dẫn đến các hạt có thể dễ dàng va chạm với bề mặt và gây tổn thương hơn so với đường dẫn khí lớn.
- Các đường thở nhỏ được xác định có đường kính < 2 mm và phát sinh từ thế hệ phân nhánh thứ 4 - 14 (lấy khí quản là thế hệ thứ nhất đến phế nang là thứ 23), nhưng trung bình phát sinh từ thế hệ thứ 8.
- Người ta ước tính 20% đường dẫn khí nhỏ có đường kính < 2 mm bao gồm các phế quản với các phần tử sụn trong thành của chúng, trong khi phần còn lại là các tiểu phế quản hoặc các ống túi phế nang.
- Bệnh đường thở nhỏ (SAD: small airway disease) là một đặc điểm chính của COPD, và đã được nghiên cứu rộng rãi trong nhiều thập kỷ. Bài báo này cung cấp một đánh giá toàn diện về bệnh lý của SAD trong COPD. Chúng tôi cung cấp tổng quan lịch sử về các nghiên cứu chính về bản chất của SAD trong COPD, và mô tả các đặc điểm bệnh lý khác nhau của SAD. Chúng tôi tập trung vào các chủ đề liên quan đến lâm sàng, bao gồm mối quan hệ giữa SAD và khí phế thũng, và SAD trong giai đoạn đầu của COPD. Chúng tôi thảo luận về các cơ chế gây ra sự tiến triển của SAD, bao gồm sự xâm nhập của vi khuẩn và các đợt cấp
2. Lịch sử nghiên cứu: những quan sát ban đầu về SAD trong COPD
- Mối liên quan giữa bệnh đường thở và khí phế thũng lần đầu tiên được thảo luận vào thế kỷ XIX. Năm 1819, René Laënnec cho rằng viêm phế quản mạn và khí phế thũng là những tình trạng cùng tồn tại [5].
- William Gairdner (1850) đã thảo luận về tác động của những thay đổi đối với đường thở xa và mối liên hệ với khí thũng [6], đặt nền tảng cho sự hiểu biết hiện tại về mối liên quan giữa SAD và khí phế thũng.
- Bằng chứng mô bệnh học của SAD trong COPD tiếp tục phát triển vào giữa thế kỷ XX; Bảng 1 liệt kê những phát hiện chính của các nghiên cứu kéo dài từ năm 1953 đến năm 1971.
Đặc điểm mô bệnh học SAD ở bệnh nhân COPD
|
Author(s)
|
Đặc điểm mô bệnh học
|
|
Spain and Kaufman (1953)
|
Viêm, xơ hóa, hẹp đường thở
|
|
Leopold and Gough (1957)
|
Viêm, hẹp đường thở, xơ hóa, tắc nghẽn
|
|
McLean (1958)
|
Giãn đường thở, viêm, nút nhày
|
|
Anderson and Foraker (1962)
|
Hẹp đường thở, mất liên kết các phế nang
|
|
Pratt et al. (1965)
|
Mất liên kết các phế nang
|
|
Hogg et al. (1968)
|
Viêm, nút nhày, tắc nghẽn đường thở
|
|
Bignon et al. (1969)
|
Viêm va hẹp đường thở
|
|
Matsuba and Thurlbeck (1971)
|
Hẹp đường thở và tắc nghẽn
|
- Một đặc điểm nổi bật của những nghiên cứu này là sự không đồng nhất của những thay đổi được báo cáo, bao gồm viêm, xơ hóa, hẹp, giãn và tắc nghẽn các tiểu phế quản. Leopold và Gough báo cáo hẹp 60% các tiểu phế quản gây khí phế thũng trung tâm tiểu thùy [8].
- McLean [11] và sau đó là Hogg et al [12] ghi nhận các nút nhầy ở đường thở nhỏ có tổn thương khí phế thũng. Tình trạng mất gắn kết phế nang, kết nối xuyên tâm với các đường dẫn khí nhỏ như nan hoa bánh xe cũng giảm ở vị trí có khí phế thũng. Điều này có thể làm giảm sự thông thoáng của đường thở và khiến đường thở dễ bị xẹp hơn khi thở ra.
- Nhìn chung, những thay đổi này có thể làm giảm luồng khí, tăng khả năng bẫy khí và do đó làm giảm dung tích thông khí.
- Năm 1965, Macklem et al đo áp lực phế quản ở bệnh nhân khí phế thũng và nhóm chứng khỏe mạnh [15]. Các đường thở nhỏ được xác định là vị trí gây tắc nghẽn luồng khí ở bệnh nhân khí phế thũng.
- Nghiên cứu mang tính bước ngoặt của Hogg, Macklem và Thurlbeck vào năm 1968 bằng cách sử dụng kỹ thuật đặt ống thông ngược dòng trong phổi đã cắt bỏ của 5 bệnh nhân nhóm chứng và 7 bệnh nhân khí phế thũng [12]. Các đường thở nhỏ (đường kính <2 mm) chiếm khoảng 25% tổng sức cản đường thở ở nhóm chứng khỏe mạnh.
- Tổng sức cản đường thở tăng lên ở bệnh nhân khí phế thũng so với nhóm chứng, chủ yếu do sức cản đường thở nhỏ tăng khoảng 40 lần. Điều này có thể được giải thích bởi sự thay đổi sức cản có mối liên quan nghịch với lũy thừa 4 của giảm bán kính đường thở [16].
- Nút đờm, viêm, xơ hóa và tắc nghẽn các tiểu phế quản nhỏ là những đặc điểm mô bệnh học chính. Các tác giả đề xuất thuật ngữ “bệnh đường thở nhỏ” để mô tả những thay đổi này.
- Mức độ không đồng nhất cao trong bệnh lý mô đã được nhận xét bởi Heppleston và Leopold [17], đề xuất cần nghiên cứu sâu hơn trên các bệnh phẩm từ các trường hợp bệnh đã được xác định, hoặc các tổn thương ban đầu trước khi phổi bị phá hủy rộng hơn.
- Mead gọi các đường dẫn khí nhỏ là vùng “yên lặng”, nơi SAD dẫn đến tắc nghẽn đường thở được xác nhận bằng đo CNHH có thể không được chú ý trong nhiều năm [19], dẫn đến chẩn đoán COPD muộn.
3. Đặc điểm mô bệnh học bệnh đường thở nhỏ trong COPD
Các nghiên cứu chi tiết bằng cách sử dụng nhiều kỹ thuật mô bệnh học và hình ảnh đã làm tăng hiểu biết của chúng ta về bản chất của SAD trong COPD. Các bất thường bệnh lý quan trọng hiện đã được xem xét.
3.1. Tái cấu trúc đường thở
- Các đường thở nhỏ có thể được coi là hệ thống dẫn khí được tạo thành bởi các tiểu phế quản màng, tiểu phế quản hô hấp và các ống phế nang [20]. Các tiểu phế quản màng bao gồm các tiểu phế quản tận được lót bởi các tế bào biểu mô trụ có lông chuyển. Các tiểu phế quản hô hấp xa hơn được lót bằng cách chuyển tiếp biểu mô trụ sang biểu mô hình khối và dẫn vào các ống phế nang và các túi phế nang với biểu mô dẹt (Hình 1) [21].
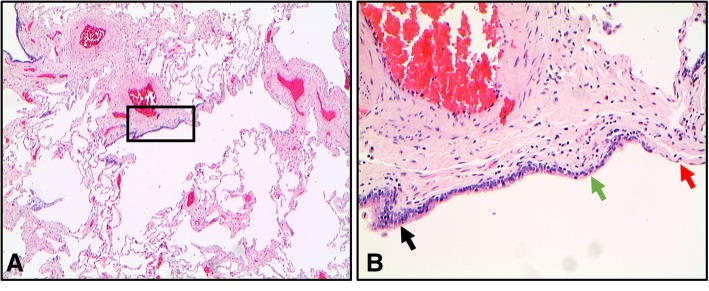
Hình 1. Chuyển tiếp cấu trúc biểu mô trong tiểu phế quản hô hấp. A. Tiểu phế quản thể hiện sự chuyển đổi từ biểu mô trụ sang biểu mô hình khối và cuối cùng là biểu mô phế nang dẹt. B Hình ảnh phóng to từ hình ảnh A cho thấy biểu mô trụ (mũi tên đen), biểu mô hình khối (mũi tên xanh) và biểu mô dẹt (mũi tên đỏ)
- Thành tiểu phế quản được chia thành các lớp: biểu mô hô hấp, màng đáy, lớp đệm niêm mạc (laminapropria), cơ trơn (giảm ở đường thở xa) và lớp ngoại mạc (adventitia).
- Không giống như thành phế quản, các tuyến thanh dịch không phải là một đặc điểm bình thường và sụn không có trong cấu trúc tiểu phế quản. Các đường thở nhỏ COPD cho thấy sự tái cấu trúc đáng kể, với độ dày của thành đường thở tăng lên so với những người hút thuốc không COPD [22]. Sự gia tăng độ dày thành phế quản bắt nguồn từ những thay đổi của biểu mô, các nút nhầy, tăng mật độ tế bào viêm, quá sản cơ trơn và xơ hóa (Hình 2).
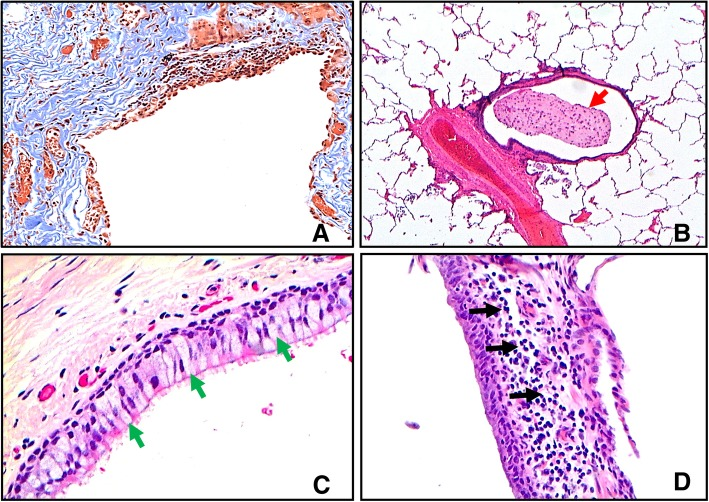
Hình 2. Đặc điểm mô bệnh học của bệnh đường thở nhỏ trong COPD. A. Tiểu phế quản COPD với thành đường thở dày lên do tái cấu trúc sợi và lắng đọng quá nhiều các bó collagen (màu xanh lam). B Một bó phế quản mạch máu COPD, trong đó tiểu phế quản chứa một nút nhầy lớn trong lòng (mũi tên đỏ). C Tiểu phế quản COPD với sự gia tăng số lượng tế bào hình đài (màu xanh lá cây) trong lớp biểu mô. D Thành của tiểu phế quản COPD với số lượng tế bào viêm tăng lên (mũi tên đen)
- Tái cấu trúc đường thở là do quá trình lành vết thương để đáp ứng với tổn thương do các tác nhân kích thích như: khói thuốc lá, vi rút, vi khuẩn gây ra. Việc lành vết thương được điều chỉnh chặt chẽ bởi sự tương tác giữa phản ứng miễn dịch với các kích thích này và việc sửa chữa và tái tạo chất nền ngoại bào của nguyên bào sợi [23].
- Nguyên bào sợi cơ (Myofibroblasts) phần lớn có nguồn gốc từ nguyên bào sợi dưới biểu mô, các tế bào biểu mô xung quanh trải qua quá trình chuyển tiếp trung mô biểu mô (EMT) và các tế bào sợi tuần hoàn. Chất nền ngoại bào (ECM) sau đó được tái cấu trúc bởi các enzym ly giải protein, chẳng hạn metalloproteinase, để thiết lập lại cân bằng nội môi của mô. Tuy nhiên, do khả năng tái tạo của mô phổi bị hạn chế, quá trình này thường để lại sẹo. Các nguyên bào sợi cơ trải qua quá trình apoptosis (chết theo chương trình) để ngăn ngừa quá trình tạo sẹo và tái cấu trúc sơ quá mức. Tuy nhiên, quá trình này có thể dẫn đến những thay đổi vĩnh viễn về cấu trúc mô [24].
- Biểu mô hô hấp bao gồm bốn loại tế bào; tế bào đáy, tế bào có lông hút, tế bào tiết và tế bào trung gian [25]. Sau chấn thương, cần tái tạo biểu mô với các loại tế bào biệt hóa để phục hồi chức năng bình thường. Tuy nhiên, tái cấu trúc bất thường biểu mô xảy ra trong COPD với những thay đổi phổ biến bao gồm chuyển sản tế bào hình đài, tăng sản tế bào đáy và chuyển sản vảy. Những thay đổi này thường xuyên hơn đường thở nhỏ COPD so với nhóm chứng [26, 27].
- Tế bào đáy hoạt động như tế bào tiền thân cho các tế bào có lông chuyển và các tế bào tiết trong quá trình biểu mô hóa [25, 28]. Hút thuốc lá làm thay đổi chương trình phiên mã của tế bào đáy, gây ra quá trình sửa chữa sai lệch [28]. EMT dường như tồn tại trong biểu mô đường thở nhỏ ở bệnh nhân COPD, các marker của nguyên bào sợi, α-actin cơ trơn và vimentin tăng lên trong các tế bào biểu mô đường thở nhỏ ở bệnh nhân COPD so với nhóm chứng không hút thuốc, và điều này tương quan nghịch với giới hạn luồng khí [29, 30].
- Những thay đổi đối với biểu mô phế quản có thể làm thay đổi tương tác giữa vật chủ và vi khuẩn. Ví dụ, khói thuốc lá điều chỉnh sự biểu hiện của thụ thể yếu tố hoạt hóa tiểu cầu, thúc đẩy sự gia tăng sự bám dính của Haemophilus influenzae (NTHi) và Streptococcus pneumoniae không thể chuyển hóa đối với các tế bào biểu mô phế quản [32]. Hơn nữa, các tế bào biểu mô phế quản COPD tạo ra một lượng thấp hơn các peptide chống vi khuẩn, human defensin-2 và S100A7, khi được đồng thời nuôi cấy với rhinovirus ở người, Pseudomonas aeruginosa hoặc NTHi so với đối chứng [33, 34].
- Ngoài ra còn có giảm globulin miễn dịch niêm mạc A (IgA) [26], các điều kiện môi trường trong đường thở nhỏ COPD dường như tạo điều kiện thuận lợi cho sự quần cư (colonisation) của vi sinh vật gây bệnh, có thể làm tăng thêm tình trạng viêm và tổn thương mô.
- Xơ hóa xảy ra trong đường thở nhỏ COPD, với sự gia tăng độ dày của các lớp niêm mạc riêng lẻ và tăng sự lắng đọng protein ECM (extracellular matrix)
- Mỗi lớp niêm mạc được cấu tạo bởi một mạng lưới các protein bao gồm collagens, laminin và proteoglycan.
- Eurlings và CS cho thấy nồng độ collagen toàn phần tăng lên đáng kể trong thành đường thở nhỏ của bệnh nhân COPD GOLD- II và GOLD- IV so với nhóm chứng hút thuốc [35].
- Tương tự, Kranenberg và CS cũng ghi nhận sự gia tăng collagens I, III và IV ở màng đáy và collagens I và III ở lớp đệm niêm mạc (lamina propria) và lớp ngoại mạc (adventitia) của bệnh nhân COPD so với nhóm chứng hút thuốc [36]. Các protein khác tăng trong niêm mạc đường dẫn khí nhỏ COPD bao gồm laminin, tenascin và fibronectin [36, 37].
3.2. Nút nhầy
- Số lượng đường dẫn khí nhỏ có nút nhầy tăng ở bệnh nhân COPD và tương ứng với mức độ nặng của bệnh [22]. Tắc các đường thở nhỏ do nút nhầy được chứng minh có liên quan đến tử vong sớm ở những bệnh nhân bị khí phế thũng nặng được điều trị bằng phẫu thuật giảm thể tích phổi [47].
- Tăng tiết chất nhầy có thể gây ra rối loạn chức năng đường thở nhỏ do tắc nghẽn luồng khí vật lý hoặc bằng cách chứa các vi sinh vật gây bệnh, thúc đẩy quá trình viêm và phá hủy mô thêm [48].
- Bản thân việc hút thuốc lá gây ra những thay đổi bệnh lý liên quan đến tăng tiết chất nhầy, phì đại tuyến nhầy, phì đại tế bào hình đài. Tăng sản ở đường thở lớn, tăng sản tế bào hình đài và chuyển sản ở đường thở nhỏ ở những người đang hút thuốc so với những người không hút thuốc [49].
- Chất nhầy là một hỗn hợp các tế bào miễn dịch, dịch tiết tế bào, muối, lipid và protein bao gồm các enzym và các chất trung gian gây viêm. Thành phần của chất nhầy thay đổi theo dấu hiệu môi trường. Điều này được minh họa bằng những thay đổi được quan sát thấy trong các đợt cấp COPD, theo đó số lượng bạch cầu trung tính trong đờm và mức độ của phối tử chemokine CXCL8, interleukin-17A (IL-17A), TNF-α và IL-1β được tăng lên so với trạng thái ổn định [50].
- Mucin là glycoprotein được tìm thấy trong chất nhầy, có đặc tính đàn hồi đóng góp vào đặc tính lý sinh của chất nhầy. Trong điều kiện bình thường, hàm lượng mucin của đờm khoảng 2-5%. Tuy nhiên, sự tăng nhỏ nồng độ của chất nhầy có thể làm thay đổi đặc tính lý sinh của đờm, khiến biểu mô lông chuyên khó di chuyển chất nhầy dọc theo đường hô hấp [51].
- Hút thuốc lá làm ngắn chiều dài lông chuyển trong đường thở nhỏ và điều này càng trở nên tồi tệ hơn ở bệnh nhân COPD. Một phần có thể do sự gia tăng thực bào lông chuyển (ciliophagy), một quá trình theo đó chiều dài lông chuyển ngắn lại để đáp ứng với việc tiếp xúc với khói thuốc lá. Tần số đập của lông mao ở bệnh nhân COPD giảm so với người khỏe mạnh hút thuốc. Chức năng của lông mao giảm sẽ làm giảm sự di chuyển của chất nhầy dọc theo đường hô hấp [52].
- Bằng chứng về mối liên quan giữa tăng tiết chất nhầy và bản thân COPD (chứ không phải hút thuốc chủ động) ít rõ ràng hơn, với một số nghiên cứu cho thấy có sự khác biệt hoặc không có sự khác biệt về số lượng tế bào hình đài trong đường thở nhỏ giữa người hút thuốc khỏe mạnh và bệnh nhân COPD [53].
- Thurlbeck và CS báo cáo những người bị cả viêm phế quản mạn và khí phế thũng có số lượng tế bào hình đài tăng lên so với nhóm chứng hút thuốc. Điều này cho thấy mức độ chuyển sản tế bào hình đài trong các đường dẫn khí nhỏ có thể liên quan đến mức độ phá hủy nhu mô [54].
- Nồng độ mucin 5AC (MUC5AC) và mucin 5B (MUC5B) trong đờm tăng ở bệnh nhân COPD so với nhóm hút thuốc khỏe mạnh và mức độ tương quan với mức độ bệnh. Hơn nữa, biểu hiện biểu mô của MUC5AC và nồng độ MUC5B trong lòng đường thở ở các tiểu phế quản không có tuyến dưới niêm mạc tăng lên ở bệnh nhân COPD so với nhóm chứng hút thuốc và không hút thuốc [55].
- Có khả năng là sự kết hợp giữa hút thuốc lá mạn tính và tiếp xúc với vi sinh vật góp phần vào cơ chế bệnh sinh SAD ở bệnh nhân COPD. Tăng chuyển sản tế bào hình đài có liên quan đến giảm nồng độ tiết IgA (SIgA) phủ trên bề mặt đường thở nhỏ ở bệnh nhân COPD. Hơn nữa, những đường thở này có mức độ RNA ribosome 16S của vi khuẩn tăng lên. SIgA rất quan trọng trong việc bảo vệ vật chủ ở bề mặt niêm mạc và do đó, sự mất mát sẽ làm tăng tính nhạy cảm với nhiễm trùng. IgA được sản xuất bởi các tế bào plasma dưới biểu mô và được vận chuyển qua biểu mô phế quản đến bề mặt trên cùng nhờ thụ thể Ig cao phân tử (pIgR) [56].
- Hai yếu tố quan trọng có thể dẫn đến việc chứa vi khuẩn gây bệnh trong đường thở COPD: giảm độ thanh thải của chất nhầy và giảm hoạt động chống vi khuẩn của chất nhầy. Những thay đổi này có thể thúc đẩy sự xâm nhập của vi khuẩn trong đường thở và làm tăng tình trạng viêm. Điều này có khả năng thúc đẩy sự tiến triển của SAD, bao gồm cả ở các giai đoạn sớm của COPD [57].
3.3. Xâm nhập tế bào miễn dịch
- Viêm đường thở nhỏ do hút thuốc dẫn đến xơ hóa và mất mô. Một số báo cáo cho thấy số lượng tế bào miễn dịch tăng thêm trong đường thở nhỏ của bệnh nhân COPD so với những người không hút thuốc và những người hút thuốc không COPD [58].
- Saetta et al ghi nhận sự gia tăng số lượng cụm biệt hóa T-CD 68+ đại thực bào, tế bào T-CD8+ trong biểu mô, tế bào T-CD8+ trong lớp đệm của đường thở nhỏ COPD so với người không hút thuốc và người hút thuốc không COPD. Một nghiên cứu sau đó cho thấy số lượng đại thực bào T-CD68+ trong biểu mô và tế bào T-CD4+ trong lớp đệm cũng cao hơn ở những bệnh nhân COPD nặng so với những bệnh nhân COPD nhẹ [59].
- Tổng số đường thở dương tính (mức độ viêm) đã được báo cáo đối với đại thực bào, bạch cầu trung tính, tế bào B-CD20+ và tế bào T-CD4+ và T-CD8+, tăng theo mức độ nặng của COPD. Ngược lại, thể tích tích lũy của tế bào T-CD8+ và chỉ tế bào B-CD20+ có liên quan đến mức độ nặng của COPD. Một số nghiên cứu khác báo cáo số lượng đại thực bào, bạch cầu trung tính, tế bào T-CD4+ và CD8+ tăng lên trong đường thở nhỏ COPD [60].
- Một cách tiếp cận thay thế là kết hợp phân tích sự xâm nhập của tế bào miễn dịch với đánh giá quá trình tái cấu trúc đường thở nhỏ. Số lượng các nang lympho liên quan đến đường thở nhỏ tăng lên theo mức độ nặng của COPD. Một yếu tố khác cần xem xét là việc sử dụng corticosteroid, có liên quan đến việc giảm số lượng nang lympho [61].
3.4. Liên quan giữa SAD và khí phế thũng
- Khí phế thũng trung tâm tiểu thùy có liên quan chặt chẽ với hút thuốc và khác biệt với khí phế thũng toàn tiểu thùy (có liên quan với sự thiếu hụt α1-antitrypsin) và khí phế thũng cạnh vách [62].
- Khí phế thũng trung tâm tiểu thùy ảnh hưởng đến các tiểu thùy phổi thứ cấp, là những đơn vị cấu trúc phổi không đều, đa diện được xác định bởi vách ngăn liên tiểu thùy và được cung cấp bởi một nhánh động mạch phổi và tiểu phế quản thế hệ trước tiểu phế quản tận. Các tiểu thùy phổi thứ cấp chứa 3 –10 chùm nang (acinus). Mỗi acinus bao gồm các tiểu phế quản hô hấp, các ống dẫn khí phế nang và các khoang phế nang [63].
- Sự phá hủy khí phế thũng thường liên quan đến việc hút thuốc lá xuất phát từ trung tâm của tiểu thùy, do đó thuật ngữ khí phế thũng trung tâm, do Leopold và Gough đề xuất lần đầu tiên vào năm 1957 [64].
- Các tiểu phế quản hô hấp xuất phát từ trung tâm của tiểu thùy phổi, và đây là những cấu trúc bị ảnh hưởng chủ yếu bởi khí phế thũng trung tâm tiểu thùy (còn gọi là trung tâm chùm nang) [65].
- Ngược lại, khí phế thũng toàn tiểu thùy ảnh hưởng đến tất cả các cấu trúc từ phế nang xa đến tiểu phế quản hô hấp, trong khi khí phế thũng cạnh vách ảnh hưởng đến phế nang và ống phế nang. Việc tái cấu trúc đường thở nhỏ, đặc biệt là xơ hóa, liên quan đến khí phế thũng do một mặt có sự dày lên của mô, mặt khác có sự mất mô [66].
- Gosselink và CS cho rằng một số đường dẫn khí nhỏ trải qua sự phá hủy tương tự như nhu mô trong khi những đường thở nhỏ khác có cấu trúc dày lên [67].
- Các nghiên cứu sử dụng chụp micro-CT cho thấy tổng số tiểu phế quản tận và tiểu phế quản hô hấp cấp một (chuyển tiếp) ở bệnh nhân COPD giảm so với nhóm chứng, có sự gia tăng mất đường thở nhỏ liên quan đến mức độ nặng của bệnh; 90% tiểu phế quản tận bị tắc nghẽn ở phổi bệnh nhân COPD giai đoạn IV [68-88].
- Koo và CS ghi nhận giảm số lượng tiểu phế quản tận và tiểu phế quản chuyển tiếp mặc dù không có khí phế thũng. Ở bệnh nhân GOLD 1, mức giảm của tiểu phế quản tận và tiểu phế quản hô hấp lần lượt là 29 và 41% và ở bệnh nhân GOLD 2, mức giảm tương ứng là 40 và 53% [89].
- Các đường dẫn khí nhỏ còn lại thành dày lên và lòng hẹp lại do các chất nhầy và chất keo lắng đọng. Việc mất và tái cấu trúc các tiểu phế quản tận và tiểu phế quản chuyển tiếp trong mô phổi không bị ảnh hưởng bởi khí phế thũng cung cấp thêm bằng chứng rằng SAD có trước các tổn thương khí phế thũng [90].
- Có lẽ thông khí bàng hệ qua các kênh nội phế quản và trong phế nang bảo vệ các vùng trao đổi khí ở xa cho đến khi những vùng này cũng không thể chống chọi lại được. Sự cân bằng của proteinase và anti-proteinase là một khái niệm cổ điển trong cơ chế bệnh sinh của COPD [90, 91].
- Đợt cấp COPD có liên quan đến tăng bạch cầu trung tính trong đờm và protease bạch cầu trung tính. Có sự gia tăng biểu hiện và hoạt động của chất nền metalloproteinase trong dịch rửa phế quản phế nang của bệnh nhân COPD trong đợt cấp, cho thấy rối loạn điều hòa hoạt tính proteinase ở phổi xa [92 – 95]. Protein có liên quan đến sự luân chuyển của ECM và do đó có liên quan chặt chẽ đến sự phá hủy mô. Chúng cũng có một vai trò trong quá trình xử lý mucin. Độ ổn định của mucin cao nhất khi bắt đầu đợt cấp COPD và điều này có liên quan đến việc giảm hoạt tính elastase của bạch cầu trung tính và tăng hoạt tính của chất ức chế alpha 1 protease. Đợt cấp COPD có gây giảm di chuyển của chất nhầy ở SAD do tăng nồng độ và độ nhớt của mucin. [96, 97, 98].
4. Hướng tiếp cận tương lai
- SAD hiện diện ở tất cả các giai đoạn của COPD, nhưng chúng ta ngày càng nhận thức rõ hơn tầm quan trọng của SAD trong các giai đoạn sớm của COPD [99].
- SAD dường như là tiền đề cho sự phát triển của khí phế thũng, và các chiến lược điều trị nhắm vào các đường thở nhỏ trong COPD có thể làm giảm tốc độ tiến triển của khí phế thũng. Những vấn đề về dược lý như vậy nên được chú trọng sớm hơn.
Chúng tôi đã thảo luận về những hiểu biết mới trong quá trình tiến triển của SAD bao gồm vai trò của vi khuẩn trong việc thúc đẩy SAD, vai trò của các đợt cấp có thể thúc đẩy quá trình tái cấu trúc và viêm có liên quan đến SAD, vai trò xâm nhập của vi khuẩn như giảm các peptid kháng khuẩn và tăng các vị trí bám dính. Hơn nữa, việc tăng ứ đọng chất nhầy (do tăng biểu hiện của protein mucin), kết hợp với giảm đặc tính kháng khuẩn của chất nhầy (giảm SIgA), tạo cơ hội lý tưởng cho vi khuẩn xâm nhập vào phổi, vốn đã bị tổn thương do hút thuốc lá, gây ra sự phá vỡ vi môi trường [99].
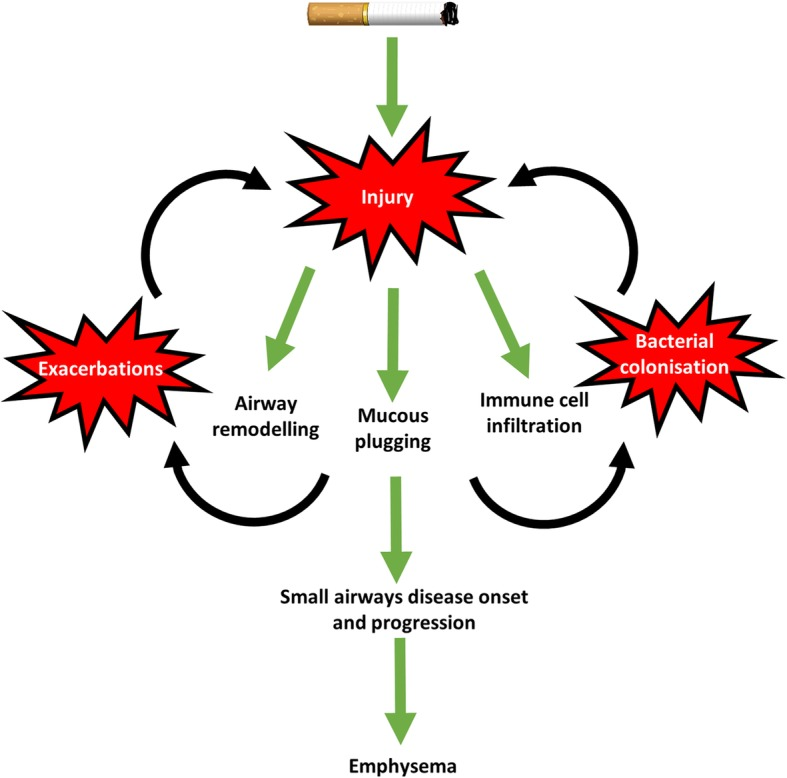
Cơ chế khởi phát và tiến triển của bệnh đường thở nhỏ trong COPD. Hút thuốc lá gây tổn thương các đường thở nhỏ. Các quá trình sửa chữa tổn thương dẫn đến tái cấu trúc đường thở quá mức, nút nhầy và xâm nhập tế bào miễn dịch. Những yếu tố này góp phần vào sự khởi phát và tiến triển của bệnh đường thở nhỏ, có trước khí phế thũng. Những thay đổi này có thể dẫn đến gia tăng đợt cấp COPD và sự xâm nhập của vi khuẩn, do đó, có thể góp phần vào sự tiến triển của bệnh đường thở nhỏ và phát triển khí phế thũng
- SAD hiện diện ở tất cả các giai đoạn của COPD, nhưng chúng ta ngày càng nhận thức rõ hơn tầm quan trọng của nó trong các giai đoạn sớm của COPD. SAD dường như là tiền đề cho sự phát triển của khí phế thũng, và các chiến lược điều trị nhắm vào các đường dẫn khí nhỏ trong COPD có thể làm giảm tốc độ tiến triển của khí phế thũng. Việc nhắm mục tiêu dược lý như vậy nên được tập trung sớm hơn trong diễn tiến tự nhiên của COPD [100].
- Tác động của sự xâm lấn của vi khuẩn có thể là viêm cấp, viêm mạn tính mức độ thấp trong một khoảng thời gian nhiều năm, dẫn đến sự xuất hiện các đợt cấp. Cả hai có thể sẽ tác động đến sự xuất hiện và tiến triển của SAD. Tổng quan này nêu bật các cơ hội tiềm năng cho các can thiệp dược lý có thể phá vỡ sự tiến triển của bệnh lý đường thở nhỏ trong COPD. Đặc biệt các can thiệp nhắm đến phục hồi khả năng phòng thủ chống vi khuẩn của đường thở nhỏ. Việc sử dụng các loại thuốc chống viêm không giải quyết được nhiễm khuẩn dai dẳng và ít có hiệu quả, vì các loại thuốc này tạo thuận lợ cho sự tồn tại của vi khuẩn có thể kích thích phản ứng miễn dịch bẩm sinh bất thường là một đặc điểm đặc trưng của COPD.
- Các mục tiêu tiềm năng bao gồm khôi phục khả năng phòng thủ chống vi khuẩn bằng cách thiết lập lại chức năng SigA, hoặc điều chỉnh các đặc tính của chất nhầy để giảm độ nhớt và cải thiện độ thanh thải nhầy, điều này cũng có thể làm giảm sự xâm nhập của vi khuẩn. Các chiến lược như vậy có thể liên quan đến việc thay đổi các đặc tính của các tế bào đáy biểu mô từ đó phục hồi chức năng nội môi và tránh sửa chữa sai lệch, bao gồm cả chuyển sản tế bào hình đài. Ngoài ra, việc điều chỉnh quá trình chữa lành vết thương để giảm các vị trí bám dính của vi khuẩn và cải thiện hoạt động diệt khuẩn của ECM vẫn có tiềm năng [101].
5. Kết luận
Nguồn gốc hiểu biết của chúng ta về mô bệnh học của SAD trong COPD có thể bắt nguồn từ đầu thế kỷ XIX. Kể từ đó, đã có rất nhiều tài liệu giúp hiểu biết thêm về các đặc điểm mô bệnh học chính của SAD. Tiến bộ trong tương lai cần tập trung vào các cơ chế phân tử thúc đẩy sự tiến triển không đồng nhất của COPD, vì người ta biết rằng một số bệnh nhân có thể tiến triển nhanh chóng trong khi những bệnh nhân khác có thể tương đối ổn định trong nhiều năm [100, 101]. Trong khi vai trò của viêm do hút thuốc lá đã được công nhận rõ ràng, thì sự đóng góp của tổn thương đối với cơ chế bảo vệ của vật chủ dẫn đến sự xâm nhập của vi khuẩn dường như rất quan trọng. Sự tác động lẫn nhau này có khả năng dẫn đến việc tái tạo và phá hủy đường thở nhỏ, đồng thời sự phát triển và tiến triển của khí thũng thứ phát sau SAD.
TS.BS. Nguyễn Quang Đợi
Trưởng khoa Nội Hô Hấp – Bệnh viện Đa khoa tỉnh Hải Dương
Ủy viên Ban chấp hành, Phó tổng thư ký Hội Hô Hấp Việt Nam
Tài liệu tham khảo
1. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Eur Respir J. 2017;49(3).
2. Stuart BO. Deposition and clearance of inhaled particles. Environ Health Perspect. 1984;55:369–90.
3. Weibel ER. Morphometry of the human lung. New York: Academic Press Inc.; 1963. p. 110–35.
4. Hogg JC. State of the art. Bronchiolitis in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(6):489–93.
5. Laennec R. Traité de l’auscultation médiate et des maladies des poumons et du coeur. New York: Samuel S and William Wood; 1819.
6. Gairdner WT. On the pathological states of the lung connected with bronchitis and bronchial obstruction. Mon J Med Sci. 1850;2(8):122–38.
7. Spain DM, Kaufman G. The basic lesion in chronic pulmonary emphysema. Am Rev Tuberc. 1953;68(1):24–30.
8. Leopold JG, Gough J. The centrilobular form of hypertrophic emphysema and its relation to chronic bronchitis. Thorax. 1957;12(3):219–35.
9. Anderson AE Jr, Foraker AG. Relative dimensions of bronchioles and parenchymal spaces in lungs from normal subjects and emphysematous patients. Am J Med. 1962;32:218–26.
10. Pratt PC, Jutabha O, Klugh GA. Quantitative relationship between structural extent of centrilobular emphysema and postmortem volume and flow characteristics of lungs. Med Thorac. 1965;22:197–209.
11. McLean KH. The pathogenesis of pulmonary emphysema. Am J Med. 1958;25(1):62–74.
12. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med. 1968;278(25):1355–60.
13. Bignon J, Khoury F, Even P, Andre J, Brouet G. Morphometric study in chronic obstructive bronchopulmonary disease. Pathologic, clinical, and physiologic correlations. Am Rev Respir Dis. 1969;99(5):669–95.
14. Matsuba K, Thurlbeck WM. The number and dimensions of small airways in nonemphysematous lungs. Am Rev Respir Dis. 1971;104(4): 516–24. 1
5. Macklem PT, Fraser RG, Brown WG. Bronchial pressure measurements in emphysema and bronchitis. J Clin Invest. 1965;44:897–905.
16. Burgel PR. The role of small airways in obstructive airway diseases. Eur Respir Rev. 2011;20(119):23–33.
17. Heppleston AG, Leopold JG. Chronic pulmonary emphysema: anatomy and pathogenesis. Am J Med. 1961;31:279–91.
18. Martinez FJ, Han MK, Allinson JP, Barr RG, Boucher RC, Calverley PMA, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(12): 1540–51.
19. Mead J. The lung’s “quiet zone”. N Engl J Med. 1970;282(23):1318–9.
20. Allen TC. Pathology of small airways disease. Arch Pathol Lab Med. 2010; 134(5):702–18.
21. Berg K, Wright JL. The pathology of chronic obstructive pulmonary disease: progress in the 20th and 21st centuries. Arch Pathol Lab Med. 2016;140(12):1423–8.
22. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–53.
23. Hutchison N, Fligny C, Duffield JS. Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. 2013;1832(7):962–71.
24. Florez-Sampedro L, Song S, Melgert BN. The diversity of myeloid immune cells shaping wound repair and fibrosis in the lung. Regeneration. 2018;5(1):3–25.
25. Crystal RG. Airway basal cells. The “smoking gun” of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(12):1355–62.
26. Polosukhin VV, Richmond BW, Du RH, Cates JM, Wu P, Nian H, et al. Secretory IgA deficiency in individual small airways is associated with persistent inflammation and remodeling. Am J Respir Crit Care Med. 2017; 195(8):1010–21.
27. Polosukhin VV, Cates JM, Lawson WE, Zaynagetdinov R, Milstone AP, Massion PP, et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(3):317–27.
28. Staudt MR, Buro-Auriemma LJ, Walters MS, Salit J, Vincent T, Shaykhiev R, et al. Airway Basal stem/progenitor cells have diminished capacity to regenerate airway epithelium in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(8):955–8.
29. Gohy ST, Hupin C, Fregimilicka C, Detry BR, Bouzin C, Gaide Chevronay H, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015;45(5):1258–72.
30. Milara J, Peiro T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–20.
31. Heijink IH, Noordhoek JA, Timens W, van Oosterhout AJ, Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(11):1439–42.
32. O’Toole RF, Shukla SD, Walters EH. Does upregulated host cell receptor expression provide a link between bacterial adhesion and chronic respiratory disease? J Transl Med. 2016;14(1):304.
33. Arnason JW, Murphy JC, Kooi C, Wiehler S, Traves SL, Shelfoon C, et al. Human beta-defensin-2 production upon viral and bacterial co-infection is attenuated in COPD. PLoS One. 2017;12(5):e0175963.
34. Amatngalim GD, Schrumpf JA, Henic A, Dronkers E, Verhoosel RM, Ordonez SR, et al. Antibacterial defense of human airway epithelial cells from chronic obstructive pulmonary disease patients induced by acute exposure to nontypeable Haemophilus influenzae: modulation by cigarette smoke. J Innate Immun. 2017;9(4):359–74.
35. Eurlings IM, Dentener MA, Cleutjens JP, Peutz CJ, Rohde GG, Wouters EF, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. 2014;14:90.
36. Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006; 126(5):725–35.
37. Annoni R, Lancas T, Yukimatsu Tanigawa R, de Medeiros Matsushita M, de Morais Fernezlian S, Bruno A, et al. Extracellular matrix composition in COPD. Eur Respir J. 2012;40(6):1362–73.
38. Simon T, Bromberg JS. Regulation of the immune system by laminins. Trends Immunol. 2017;38(11):858–71.
39. Chen J, Carcamo JM, Borquez-Ojeda O, Erdjument-Bromage H, Tempst P, Golde DW. The laminin receptor modulates granulocyte-macrophage colony-stimulating factor receptor complex formation and modulates its signaling. Proc Natl Acad Sci U S A. 2003;100(24):14000–5.
40. Chiba K, Zhao W, Chen J, Wang J, Cui HY, Kawakami H, et al. Neutrophils secrete MIP-1 beta after adhesion to laminin contained in basement membrane of blood vessels. Br J Haematol. 2004;127(5):592–7.
41. Adair-Kirk TL, Atkinson JJ, Kelley DG, Arch RH, Miner JH, Senior RM. A chemotactic peptide from laminin alpha 5 functions as a regulator of inflammatory immune responses via TNF alpha-mediated signaling. J Immunol. 2005;174(3):1621–9.
42. Khan KM, Falcone DJ. Role of laminin in matrix induction of macrophage urokinase-type plasminogen activator and 92-kDa metalloproteinase expression. J Biol Chem. 1997;272(13):8270–5.
43. Jalalvand F, Su YC, Morgelin M, Brant M, Hallgren O, Westergren-Thorsson G, et al. Haemophilus influenzae protein F mediates binding to laminin and human pulmonary epithelial cells. J Infect Dis. 2013;207(5):803–13.
44. Tan TT, Forsgren A, Riesbeck K. The respiratory pathogen moraxella catarrhalis binds to laminin via ubiquitous surface proteins A1 and A2. J Infect Dis. 2006;194(4):493–7.
45. Singh B, Alvarado-Kristensson M, Johansson M, Hallgren O, WestergrenThorsson G, Morgelin M, et al. The respiratory pathogen Moraxella catarrhalis targets collagen for maximal adherence to host tissues. MBio. 2016;7(2):e00066.
46. Abdillahi SM, Bober M, Nordin S, Hallgren O, Baumgarten M, Erjefalt J, et al. Collagen VI is upregulated in COPD and serves both as an adhesive target and a bactericidal barrier for Moraxella catarrhalis. J Innate Immun. 2015; 7(5):506–17.
47. Hogg JC, Chu FS, Tan WC, Sin DD, Patel SA, Pare PD, et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am J Respir Crit Care Med. 2007;176(5):454–9.
48. Cosio MG, Hale KA, Niewoehner DE. Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am Rev Respir Dis. 1980;122(2):265–21.
49. Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC, et al. Epithelial mucin stores are increased in the large airways of smokers with airflow obstruction. Chest. 2006;130(4):1102–8.
50. Lumsden AB, McLean A, Lamb D. Goblet and Clara cells of human distal airways: evidence for smoking induced changes in their numbers. Thorax. 1984;39(11):844–9.
51. Ebert RV, Terracio MJ. The bronchiolar epithelium in cigarette smokers. Observations with the scanning electron microscope. Am Rev Respir Dis. 1975;111(1):4–11.
52. Zheng Z, Qi Y, Xu X, Jiang H, Li Z, Yang Q, et al. Sputum mucin 1 is increased during the acute phase of chronic obstructive pulmonary disease exacerbation. J Thorac Dis. 2017;9(7):1873–82.
53. Damera G, Pham TH, Zhang J, Ward CK, Newbold P, Ranade K, et al. A sputum proteomic signature that associates with increased IL-1beta levels and bacterial exacerbations of COPD. Lung. 2016;194(3):363–9.
54. Roos AB, Sethi S, Nikota J, Wrona CT, Dorrington MG, Sanden C, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(4):428–37.
55. Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61(2):86–100.
56. Hessel J, Heldrich J, Fuller J, Staudt MR, Radisch S, Hollmann C, et al. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS One. 2014;9(1):e85453.
57. Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–30.
58. Yaghi A, Zaman A, Cox G, Dolovich MB. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir Med. 2012; 106(8):1139–47.
59. Saetta M, Turato G, Baraldo S, Zanin A, Braccioni F, Mapp CE, et al. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers Higham et al. Respiratory Research (2019) 20:49 Page 10 of 11 with both symptoms of chronic bronchitis and chronic airflow limitation. Am J Respir Crit Care Med. 2000;161(3 Pt 1):1016–21.
60. Caramori G, Di Gregorio C, Carlstedt I, Casolari P, Guzzinati I, Adcock IM, et al. Mucin expression in peripheral airways of patients with chronic obstructive pulmonary disease. Histopathology. 2004;45(5):477–84.
61. Thurlbeck WM, Malaka D, Murphy K. Goblet cells in the peripheral airways in chronic bronchitis. Am Rev Respir Dis. 1975;112(1):65–9.
62. Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, et al. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med. 2017; 377(10):911–22.
63. Kirkham S, Kolsum U, Rousseau K, Singh D, Vestbo J, Thornton DJ. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(10): 1033–9.
64. He SH, Zheng J, Duan MK. Induction of mucin secretion from human bronchial tissue and epithelial cells by rhinovirus and lipopolysaccharide. Acta Pharmacol Sin. 2004;25(9):1176–81.
65. Inoue D, Yamaya M, Kubo H, Sasaki T, Hosoda M, Numasaki M, et al. Mechanisms of mucin production by rhinovirus infection in cultured human airway epithelial cells. Respir Physiol Neurobiol. 2006;154(3): 484–99.
66. Zhu L, Lee PK, Lee WM, Zhao Y, Yu D, Chen Y. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am J Respir Cell Mol Biol. 2009;40(5):610–9.
67. Komatsu K, Jono H, Lim JH, Imasato A, Xu H, Kai H, et al. Glucocorticoids inhibit nontypeable Haemophilus influenzae-induced MUC5AC mucin expression via MAPK phosphatase-1-dependent inhibition of p38 MAPK. Biochem Biophys Res Commun. 2008;377(3):763–8.
68. Araki N, Yanagihara K, Morinaga Y, Yamada K, Nakamura S, Yamada Y, et al. Azithromycin inhibits nontypeable Haemophilus influenzae-induced MUC5AC expression and secretion via inhibition of activator protein-1 in human airway epithelial cells. Eur J Pharmacol. 2010;644 (1–3):209–14.
69. Yan F, Li W, Jono H, Li Q, Zhang S, Li JD, et al. Reactive oxygen species regulate Pseudomonas aeruginosa lipopolysaccharide-induced MUC5AC mucin expression via PKC-NADPH oxidase-ROS-TGF-alpha signaling pathways in human airway epithelial cells. Biochem Biophys Res Commun. 2008;366(2):513–9.
70. Ganesan S, Comstock AT, Kinker B, Mancuso P, Beck JM, Sajjan US. Combined exposure to cigarette smoke and nontypeable Haemophilus influenzae drives development of a COPD phenotype in mice. Respir Res. 2014;15:11.
71. Herr C, Han G, Li D, Tschernig T, Dinh QT, Beisswenger C, et al. Combined exposure to bacteria and cigarette smoke resembles characteristic phenotypes of human COPD in a murine disease model. Exp Toxicol Pathol. 2015;67(3):261–9.
72. Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, et al. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(1):185–94.
73. Du RH, Richmond BW, Blackwell TS Jr, Cates JM, Massion PP, Ware LB, et al. Secretory IgA from submucosal glands does not compensate for its airway surface deficiency in chronic obstructive pulmonary disease. Virchows Arch. 2015;467(6):657–65.
74. Ladjemi MZ, Lecocq M, Weynand B, Bowen H, Gould HJ, Van Snick J, et al. Increased IgA production by B-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur Respir J. 2015;45(4):980–93.
75. Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. N Engl J Med. 1974;291:755–8.
76. Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 Pt 1):822–6.
77. Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghe B, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med. 2002;166(1):105–10.
78. Eapen MS, McAlinden K, Tan D, Weston S, Ward C, Muller HK, et al. Profiling cellular and inflammatory changes in the airway wall of mild to moderate COPD. Respirology. 2017;22(6):1125–32.
79. Isajevs S, Taivans I, Svirina D, Strazda G, Kopeika U. Patterns of inflammatory responses in large and small airways in smokers with and without chronic obstructive pulmonary disease. Respiration. 2011;81(5):362–71.
80. Pilette C, Colinet B, Kiss R, Andre S, Kaltner H, Gabius HJ, et al. Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur Respir J. 2007;29(5):914–22.
81. Olloquequi J, Ferrer J, Montes JF, Rodriguez E, Montero MA, Garcia-Valero J. Differential lymphocyte infiltration in small airways and lung parenchyma in COPD patients. Respir Med. 2010;104(9):1310–8.
82. Baraldo S, Turato G, Badin C, Bazzan E, Beghe B, Zuin R, et al. Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax. 2004;59(4):308–12.
83. Battaglia S, Mauad T, van Schadewijk AM, Vignola AM, Rabe KF, Bellia V, et al. Differential distribution of inflammatory cells in large and small airways in smokers. J Clin Pathol. 2007;60(8):907–11.
84. Kim V, Kelemen SE, Abuel-Haija M, Gaughan JP, Sharafkaneh A, Evans CM, et al. Small airway mucous metaplasia and inflammation in chronic obstructive pulmonary disease. COPD. 2008;5(6):329–38.
85. Plumb J, Smyth LJ, Adams HR, Vestbo J, Bentley A, Singh SD. Increased Tregulatory cells within lymphocyte follicles in moderate COPD. Eur Respir J. 2009;34(1):89–94.
86. Hogg JC, Pare PD, Hackett TL. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol Rev. 2017;97(2):529–52.
87. Gosselink JV, Hayashi S, Elliott WM, Xing L, Chan B, Yang L, et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(12):1329–35.
88. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–75.
89. Koo HK, Vasilescu DM, Booth S, Hsieh A, Katsamenis OL, Fishbane N, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med. 2018;6(8):591–602.
90. Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(2):253–68.
91. Polverino F, Rojas-Quintero J, Wang X, Petersen H, Zhang L, Gai X, et al. A disintegrin and metalloproteinase domain-8: a novel protective proteinase in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018; 198(10):1254–67.
92. Wang X, Polverino F, Rojas-Quintero J, Zhang D, Sanchez J, Yambayev I, et al. A disintegrin and a metalloproteinase-9 (ADAM9): a novel proteinase culprit with multifarious contributions to COPD. Am J Respir Crit Care Med. 2018;198:1500–18.
93. Wedzicha JA, Calverley PMA, Albert RK, Anzueto A, Criner GJ, Hurst JR, et al. Prevention of COPD exacerbations: a European Respiratory Society/ American Thoracic Society guideline. Eur Respir J. 2017;50(3):1602265.
94. Sand JM, Martinez G, Midjord AK, Karsdal MA, Leeming DJ, Lange P. Characterization of serological neo-epitope biomarkers reflecting collagen remodeling in clinically stable chronic obstructive pulmonary disease. Clin Biochem. 2016;49(15):1144–51.
95. Schumann DM, Leeming D, Papakonstantinou E, Blasi F, Kostikas K, Boersma W, et al. Collagen degradation and formation are elevated in exacerbated COPD compared to stable disease. Chest. 2018;154:798–807.
96. Mallia-Milanes B, Dufour A, Philp C, Solis N, Klein T, Fischer M, et al. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am J Phys Lung Cell Mol Phys. 2018;315:1003–14.
97. Papakonstantinou E, Karakiulakis G, Batzios S, Savic S, Roth M, Tamm M, et al. Acute exacerbations of COPD are associated with significant activation of matrix metalloproteinase 9 irrespectively of airway obstruction, emphysema and infection. Respir Res. 2015;16:78.
98. Chillappagari S, Preuss J, Licht S, Muller C, Mahavadi P, Sarode G, et al. Altered protease and antiprotease balance during a COPD exacerbation contributes to mucus obstruction. Respir Res. 2015;16:85.
99. Shaykhiev R. Emerging biology of persistent mucous cell hyperplasia in COPD. Thorax. 2019;74(1):4–6.
100. Lange P, Celli B, Agusti A, Boje Jensen G, Divo M, Faner R, et al. Lungfunction trajectories leading to chronic obstructive pulmonary disease. N Engl J Med. 2015;373(2):111–22.
101. Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–92.